Bioanalytical Research

Bioanalytical Research Group
Mass Spectrometric Methods Development
In 1998 Matthias Mann took up a professorship in Odense, Denmark and Matthias Wilm became group leader at the EMBL. Being an instrumentation group we continued to develop methods to improve protein characterization by mass spectrometry.
Method development:
• Noise filtering technique to improve peptide detection
• Data processing to improve automated peptide identification
• Differential scanning to improve de-novo sequencing
• Parallel fragmentation of peptides to improve specific detection of secondary modifications
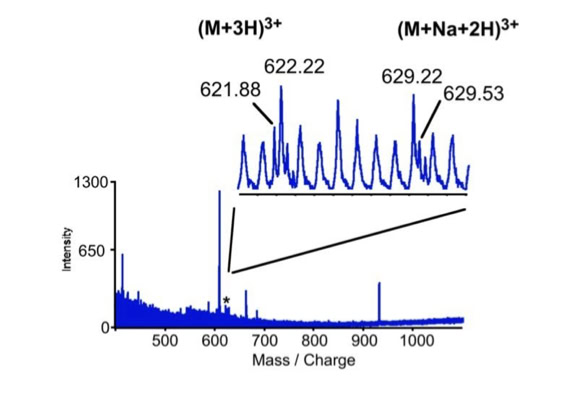
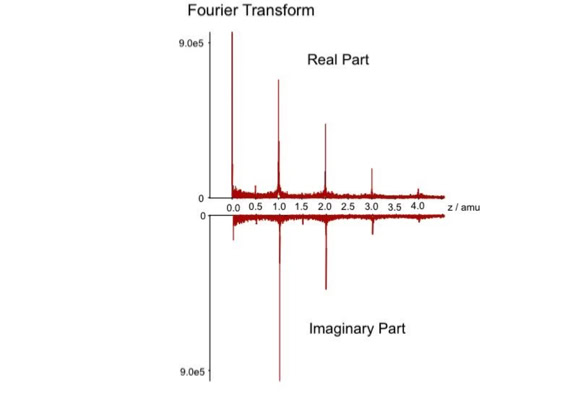
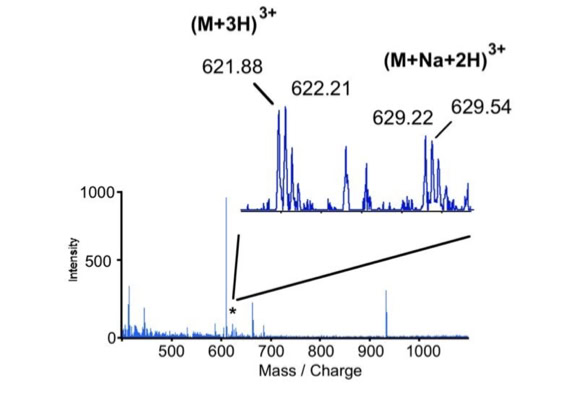
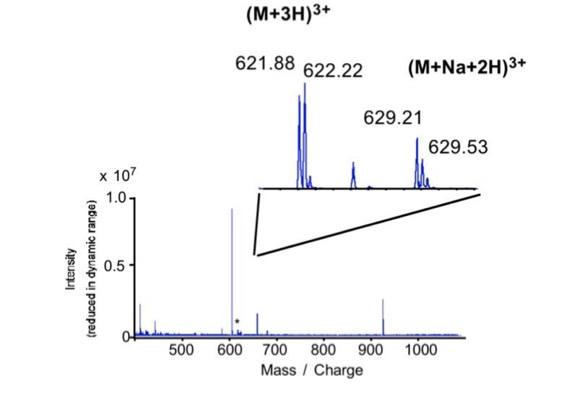
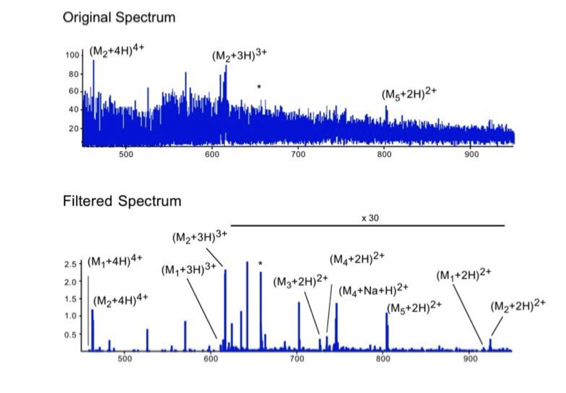
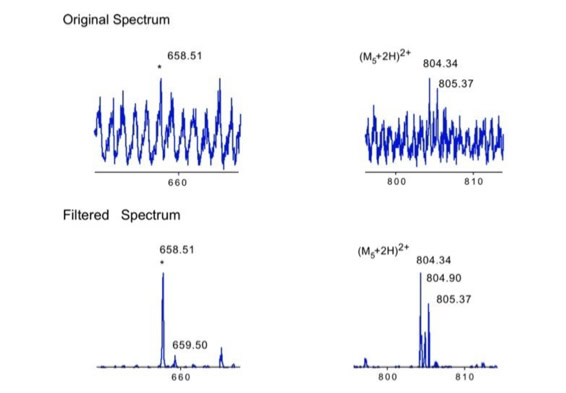
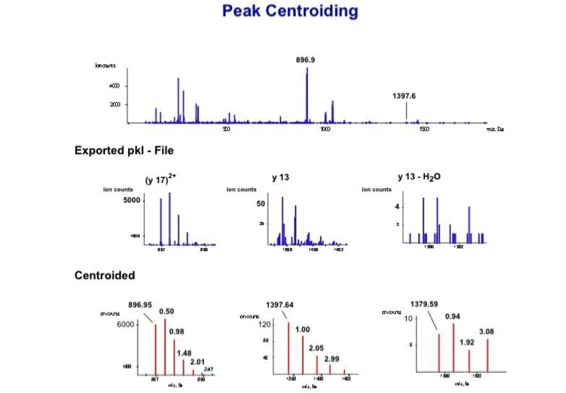
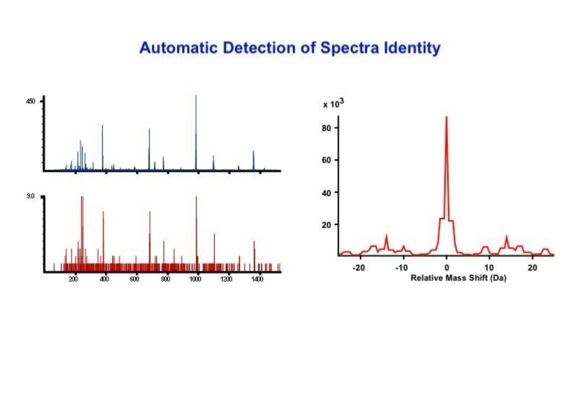
Noise filtering technique to improve peptide detection:
When sequencing peptides with the nano-electrospray ion source they are selected manually. On triple quadrupole mass spectrometers the peptide’s signal to noise can be enhanced by using precursor ion scans. This option is not available to the same degree on quadrupole time of flight machines. For these machines we developed a novel noise filtering technique to make low level peptides better visible (slide 1 - 6). In a first step regular noise is filtered by removing intensive peaks in the Fourier Transform of the spectrum. In a second step the real peptide signals are enhanced by auto-correlation [1].
Data processing to improve automated peptide identification:
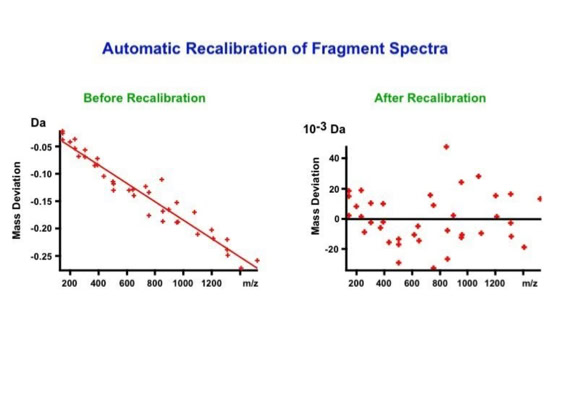
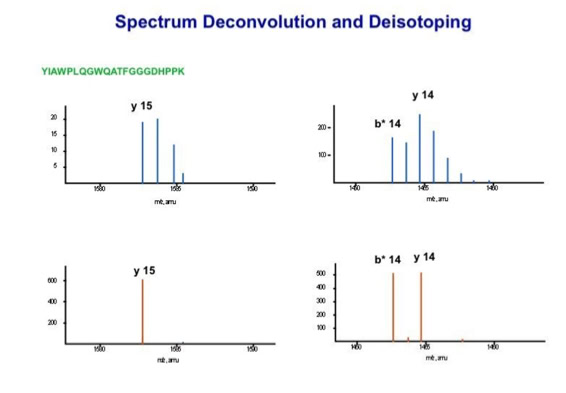
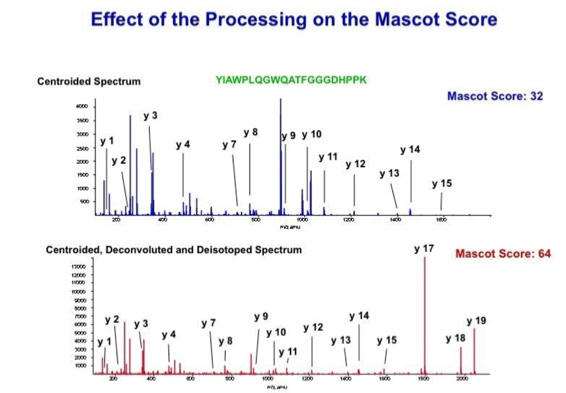
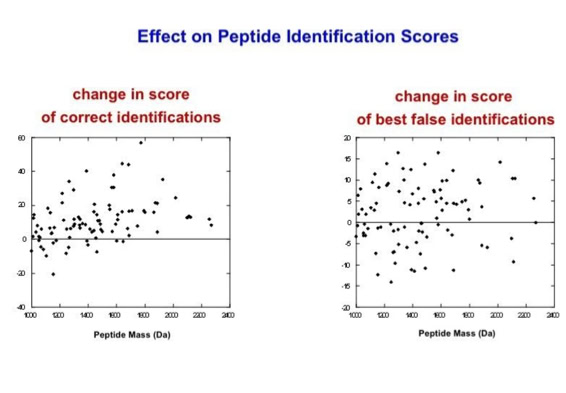
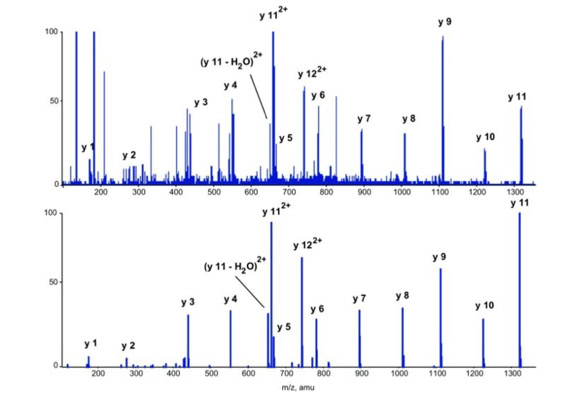
With the switch to automated data acquisition and peptide identification came the need to develop software algorithms that would help in generating the best possible specificity in data base identifications. We used a quadrupole time of flight instruments to acquire the data and Mascot as search engine. To support the identification process we pre-processed the fragment spectra (slide 7 - 10). This improved remarkably the score for correctly identified peptides whereas the score for wrongly assigned peptides stayed nearly the same (slide 11, 12) [2]. The processing was implemented in form of scripts using Igor Pro as processing engine. Later Matrix Science made a similar programme publicly accessible (Mascot Distiller).
Differential scanning to improve de novo sequencing:
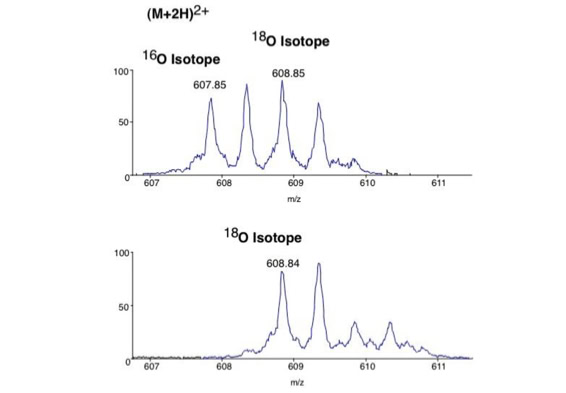
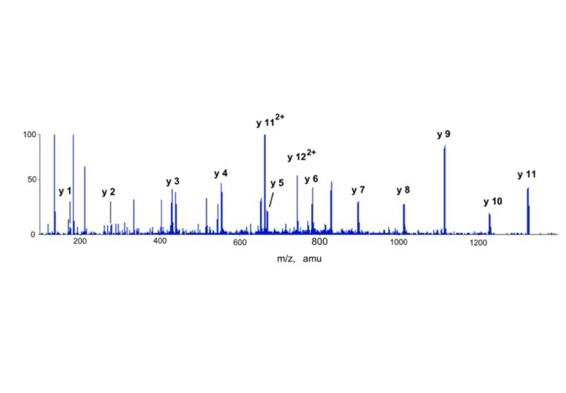
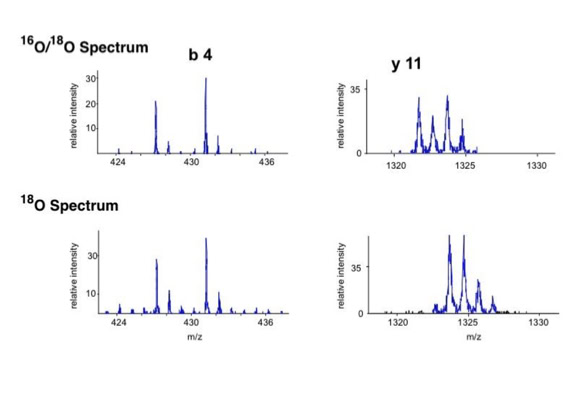
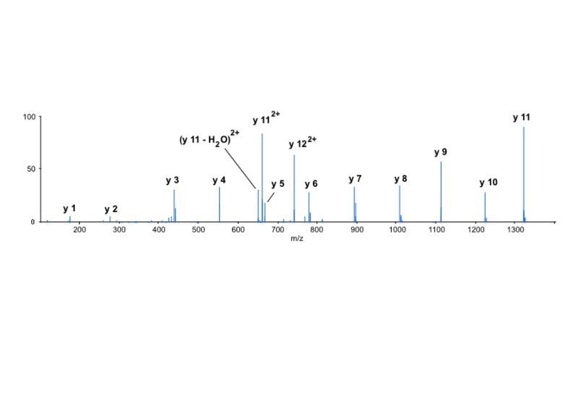
Differential scanning builds upon 18O labeling for de novo sequencing [3]. By digesting proteins in 1:1 18O/16O water all cleaved peptides carry to 50% an 18O isotope at their C-terminus. For the differential scanning method two fragment spectra per peptide are acquired, one including the entire isotopic envelope and a second with only the 18O isotopes selected (slide 13). When comparing these two fragment spectra C-terminal fragments differ by their isotopic representation (slide 15). This difference can be used by a software algorithm to generate a fragment spectrum filtered for C-terminal fragments (slide 16, 17) [4].
Parallel fragmentation of peptides to improve specific detection of secondary modifications:
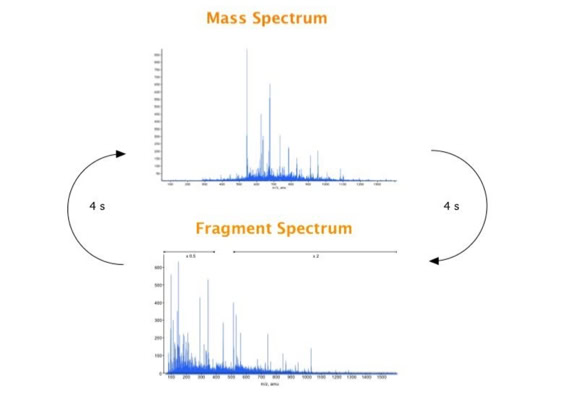
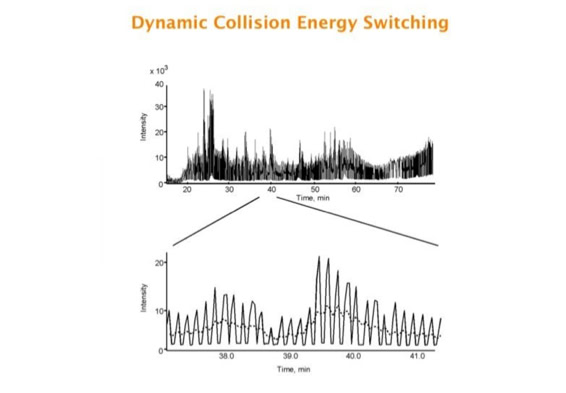
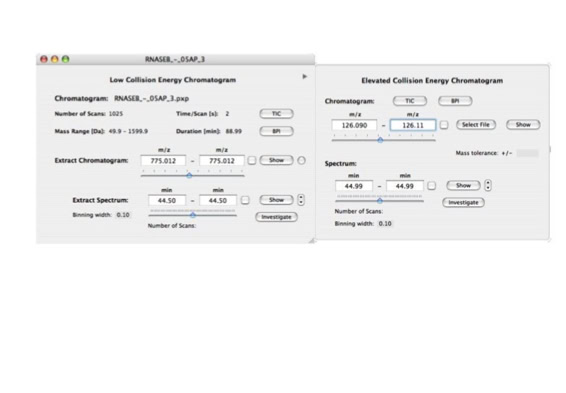
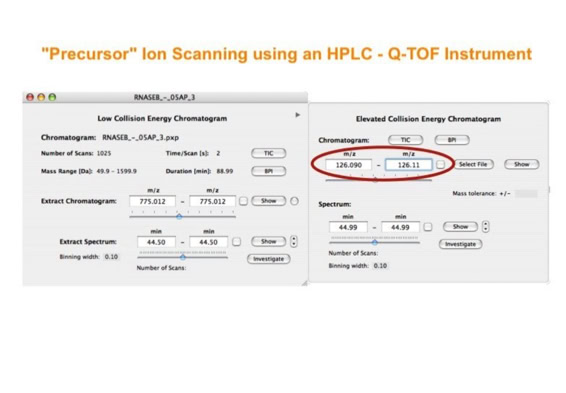
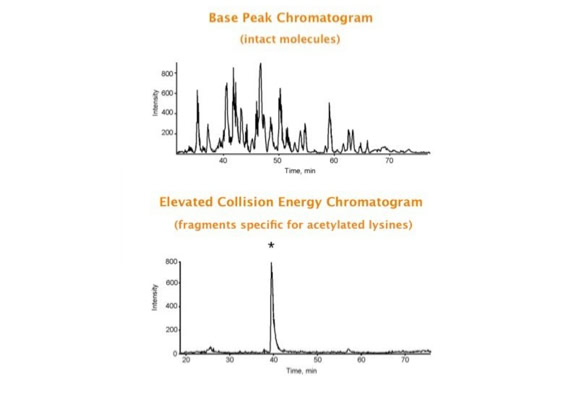
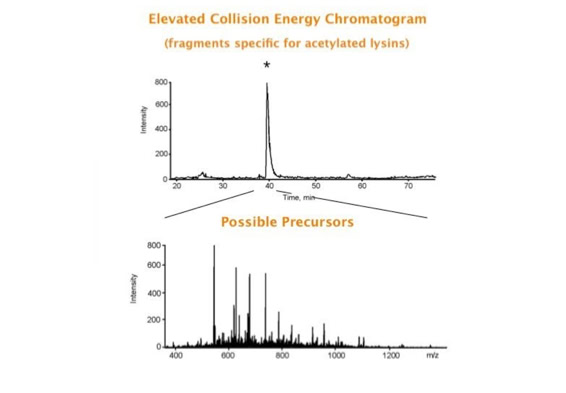
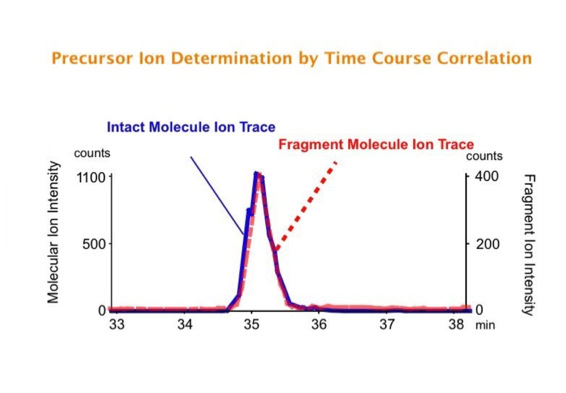
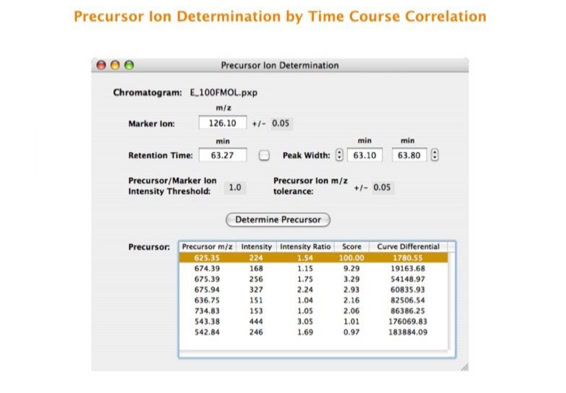
Parallel fragmentation of peptides is an attempt to increase the number of ions studied by their fragment spectrum. We explored this technique to acquire data that can be interrogated for many different marker ions or combinations of marker ions representing specific chemical structures like secondary peptide modifications. Instead of selecting an individual precursor the collision energy is permanently switched between an elevated and a low state. The mass spectrometer acquires intact masses and fragments of all the currently present molecules in an alternating way (slide 18, 19). Fragment data and intact molecular mass data can be separated by appropriate software tools (slide 20). Once the fragment data are isolated they can be interrogated for any marker ion trace or any combination of marker ions (slide 18). Once a marker ion specific for a peptide modification has been detected the true precursor that generated this marker ion upon fragmentation can be identified amongst all present precursors (slide 23) by correlating its chromatographic time profile to the time profile of the fragment (slide 24, 25). The real sequence of the precursor ion can be determined by running the same sample a second time and specifically select the highly scored precursors for fragmentation ignoring all the others [5].
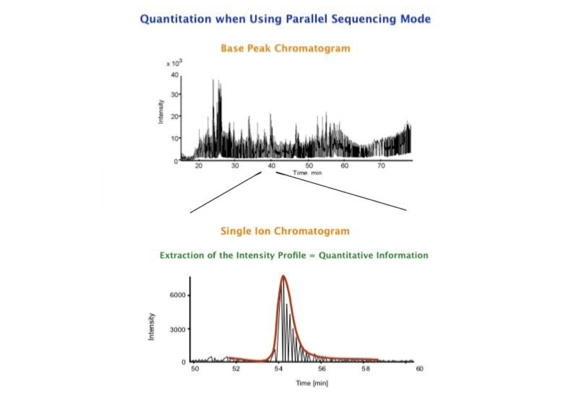
The fast switching of the mass spectrometer allows to reconstruct all chromatograms of all primary ions. The complete chromatogram gives access to their total ion volume and with it to their quantity (slide 26) [6].
The time course correlation is one of the critical steps in this analytical strategy. New equipment increases this time resolution - either by an improved LC separation using ultra high pressure chromatography equipment (Waters) or by using ion drift instruments (Prof. Clemmer). Once the time resolution is of a sufficient degree entire fragment spectra can be reconstructed for the observed precursors (parallel sequencing). The second LC run becomes superfluous.
The advantages of parallel sequencing for the analysis of very complex mixtures can be considerable. As demonstrated by scientists at Waters, qualitative and quantitative information can be generated on a systematic basis from every analysis. Prof. Clemmer's group demonstrated that it is possible to analyze proteomic samples much more rapidly covering a large number of peptides [7-10]. Since all masses are measured with time of flight instruments the high mass accuracy allows sufficiently accurate database searches.
References*
- Kast, J., M. Gentzel, M. Wilm, and K. Richardson, Noise filtering techniques for electrospray quadrupole time of flight mass spectra. J Am Soc Mass Spectrom, 2003. 14(7): p. 766-76.
- Gentzel, M., T. Kocher, S. Ponnusamy, and M. Wilm, Preprocessing of tandem mass spectrometric data to support automatic protein identification. Proteomics, 2003. 3(8): p. 1597-610.
- Shevchenko, A., Chernushevich, I., Ens, W., Standing, K. G., Thomson, B., Wilm, M., and Mann, M. (1997). Rapid 'de Novo' Peptide Sequencing by a Combination of Nanoelectrospray, Isotopic Labeling and a Quadrupole/Time-of flight Mass Spectrometer. Rapid Communications in Mass Spectrometry 11, 1015 - 1024.
- Uttenweiler-Joseph, S., G. Neubauer, S. Christoforidis, M. Zerial, and M. Wilm, Automated de novo sequencing of proteins using the differential scanning technique. Proteomics, 2001. 1(5): p. 668-682.
- Niggeweg, R., T. Kocher, M. Gentzel, A. Buscaino, M. Taipale, A. Akhtar, and M. Wilm, A general precursor ion-like scanning mode on quadrupole-TOF instruments compatible with chromatographic separation. Proteomics, 2006. 6(1): p. 41-53.
- Silva, J.C., M.V. Gorenstein, G.Z. Li, J.P. Vissers, and S.J. Geromanos, Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol Cell Proteomics, 2005.
- Hoaglund-Hyzer, C.S., J. Li, and D.E. Clemmer, Mobility labeling for parallel CID of ion mixtures. Anal Chem, 2000. 72(13): p. 2737-40.
- Moon, M.H., S. Myung, M. Plasencia, A.E. Hilderbrand, and D.E. Clemmer, Nanoflow LC/ion mobility/CID/TOF for proteomics: analysis of a human urinary proteome. J Proteome Res, 2003. 2(6): p. 589-97.
- Liu, X., M. Plasencia, S. Ragg, S.J. Valentine, and D.E. Clemmer, Development of high throughput dispersive LC-ion mobility-TOFMS techniques for analysing the human plasma proteome. Brief Funct Genomic Proteomic, 2004. 3(2): p. 177-86.
- Valentine, S.J., X. Liu, M.D. Plasencia, A.E. Hilderbrand, R.T. Kurulugama, S.L. Koeniger, and D.E. Clemmer, Developing liquid chromatography ion mobility mass spectometry techniques. Expert Rev Proteomics, 2005. 2(4): p. 553-65.
* It is not intended that the reference list reflects the entire research in the field.