Bioanalytical Research
Bioanalytical Research Group
Research Activities - EMBL


The European Molecular Biology Laboratory (EMBL) is a multi-national organisation whose primary purpose is to foster research in molecular biology. It has several outstations in European countries. In 1992 a mass spectrometry group under the heading of Matthias Mann was established to develop the use of mass spectrometry in biological research.
© 2018 Matthias Wilm Contact Me
Supportive Activity

CellZome - EMBL's functional proteomics spin-off company
General Support for Biological Research:
We continuously supported biological research at the EMBL and outside according to our capacity.
Examples:
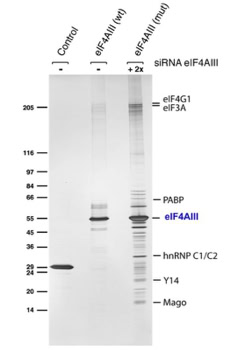
• quantitative profiling of sarcomere associated proteins in limp and extraocular muscle (slide 10) [1, 2]
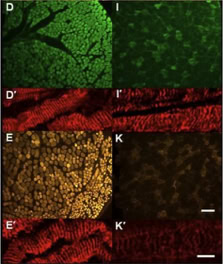
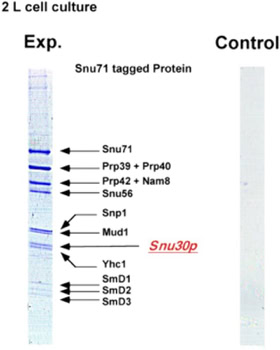
• studying the splicing mechanism by protein complex purification and sequencing at different stages of the splicing process [3]
• Nup155’s role in nuclear envelope and nuclear pore formation [4]
Proteomics Visitor Laboratory:
In 2001 we spent a considerable amount of time to establish the proteomics visitor laboratory sponsored by Waters and Biorad (slide 1-9). This visitor laboratory evolved into EMBL’s proteomics core facility.
Spin-Off Companies:
Two spin-off companies are directly related to main projects of our group, Protana, which later split off Proxeon, and CellZome.
References
• Fraterman, S., U. Zeiger, T.S. Khurana, M. Wilm, and N.A. Rubinstein, Quantitative proteomic profiling of sarcomere associated proteins in limb and extraocular muscle allotypes. Mol Cell Proteomics, 2007.
Functional Proteomics
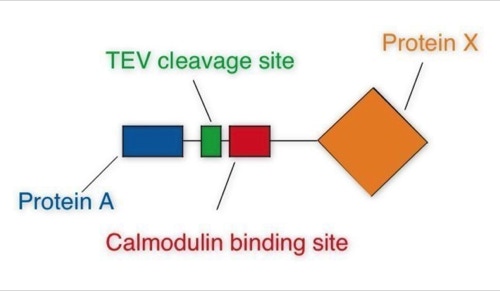
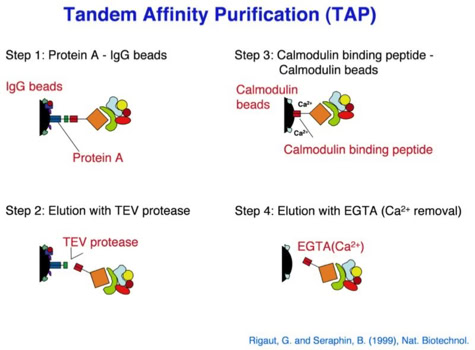
One of the early important decisions in 1999 was to dedicate all available resources to the support of the “Tandem Affinity Purification” (TAP) method [1]. The TAP method had been developed in Bertrand Seraphin’s group. It was a tag based, gentle, specific and in-vivo protein complex purification method established in yeast cells (slide 2, 3). This technology could evolve together with mass spectrometry based protein identification into a key component of an organism wide functional proteomics projects - a systematic effort to place proteins into their functional cellular context (slide 4, 6) [2].
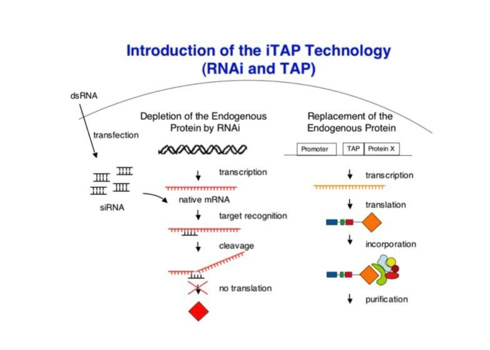
Later we demonstrated how the TAP method could be used with cells from higher eukaryotes, the iTAP project (slide 7, 8, 9) [3].
The Tandem Affinity Purification Method:
The TAP method was so important because it allowed to purify non-covalent protein complexes in a systematic fashion with high specificity without requiring individual antibodies for every complex (slide 1, 2, 3). Protein complexes are assemblies of proteins who cooperate to fulfil a specific function within the cell. At a time when the human genome was going to be published this technique would allow to place many of the newly discovered proteins into their functional context. This activity was supported by the German Bundesministerium für Bildung und Forschung (BMBF) (BioFuture, Grant 0311862 / 5).
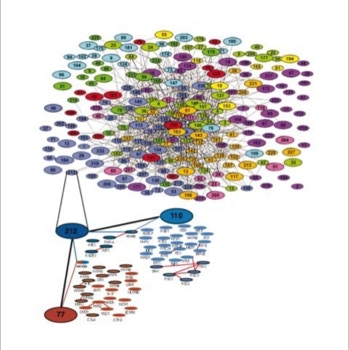
The success of this project at the EMBL motivated a proteomics spin-off company, CellZome, to use it as their core activity (slide 5, 6). They could generate the first proteome wide protein complex network [4].
Using the TAP Method in Cells from Higher Eukaryotes - the iTAP Project:
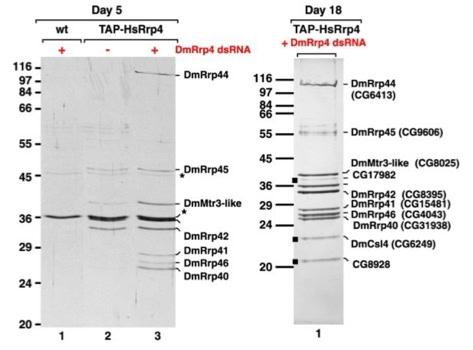
The first attempts to use TAP in Drosophila cells were a failure. Expressing a tagged protein and extracting it was often not enough to allow a specific and efficient purification of its interacting partners. This caused us to modify the experiments. Expression of the TAP-tagged protein was accompanied by suppression of its native form using interference RNA (RNAi). RNAi is a method that allows to hydrolize the native messenger RNA of a protein so that it can not be synthesized (slide 7). With the native protein missing the tagged one should be built into the biological pathways. Upon purification its native partners should be extractable. The biochemical experiments were done in the group of Elisa Izaurralde. For the protein complexes purified the project was a complete success (slide 8, 9) [3]. The iTAP method was incorporated into a patent.
iTAP Methods - Experimental Information:
References*
* It is not intended that the reference list reflects the entire research in the field.
Mass Spectrometric Methods Development
In 1998 Matthias Mann took up a professorship in Odense, Denmark and Matthias Wilm became group leader at the EMBL. Being an instrumentation group we continued to develop methods to improve protein characterization by mass spectrometry.
Method development:
• Noise filtering technique to improve peptide detection
• Data processing to improve automated peptide identification
• Differential scanning to improve de-novo sequencing
• Parallel fragmentation of peptides to improve specific detection of secondary modifications
Noise filtering technique to improve peptide detection:
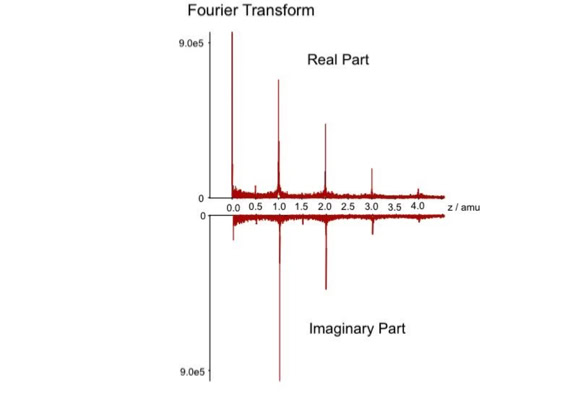
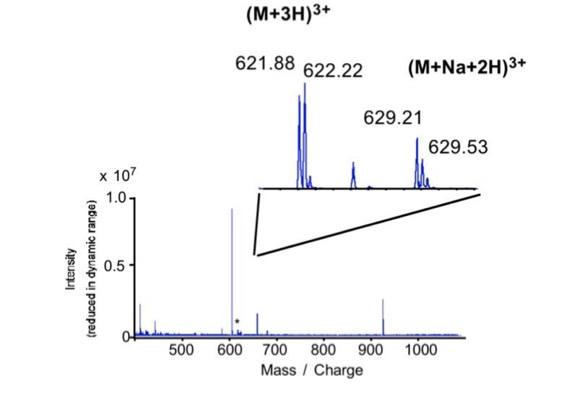
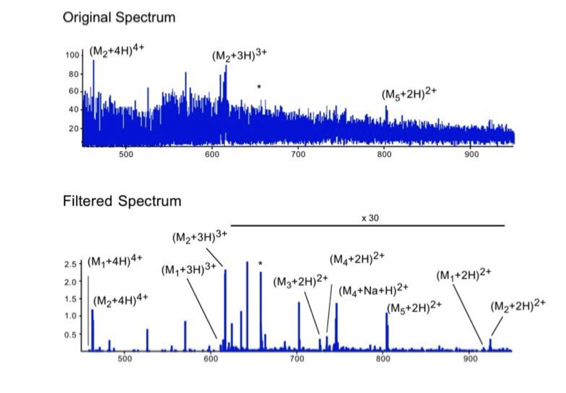
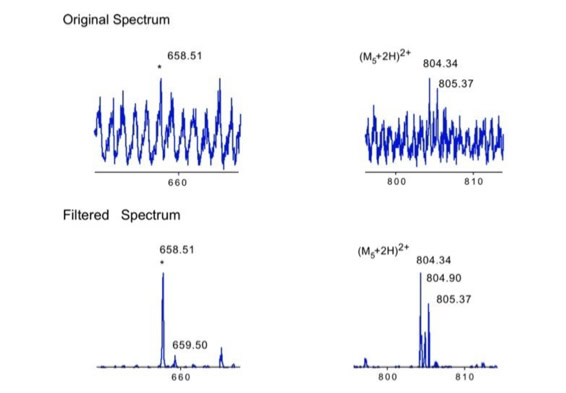
When sequencing peptides with the nano-electrospray ion source they are selected manually. On triple quadrupole mass spectrometers the peptide’s signal to noise can be enhanced by using precursor ion scans. This option is not available to the same degree on quadrupole time of flight machines. For these machines we developed a novel noise filtering technique to make low level peptides better visible (slide 1 - 6). In a first step regular noise is filtered by removing intensive peaks in the Fourier Transform of the spectrum. In a second step the real peptide signals are enhanced by auto-correlation [1].
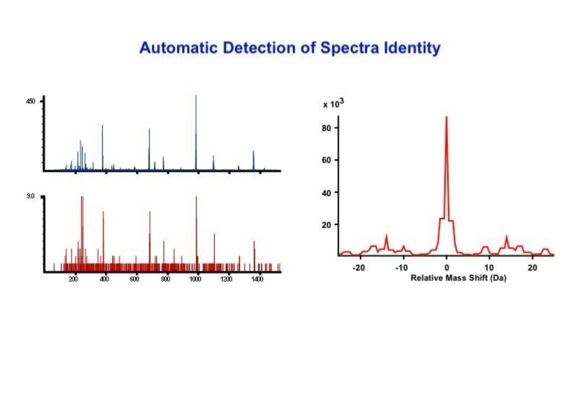
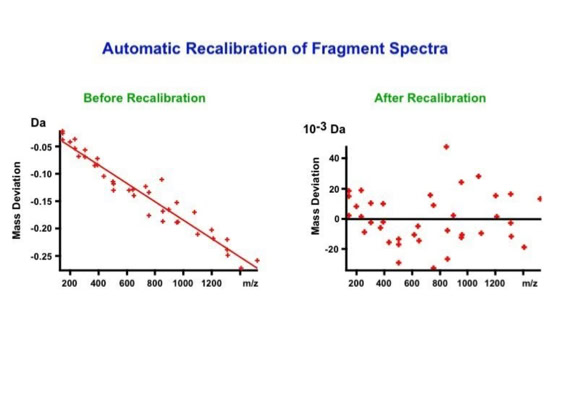
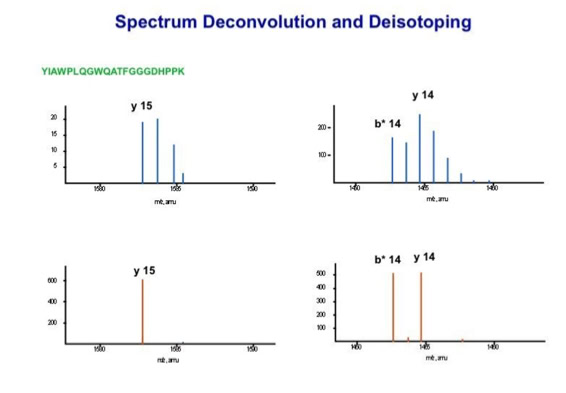
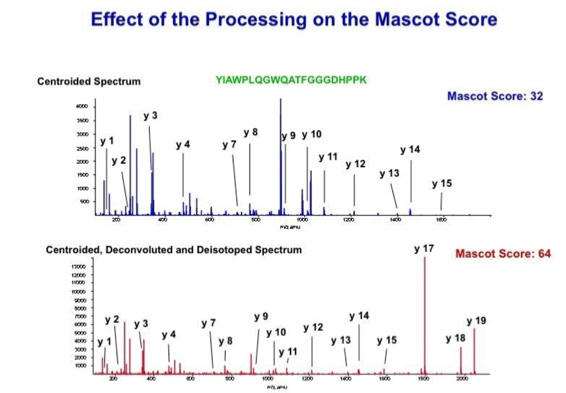
Data processing to improve automated peptide identification:
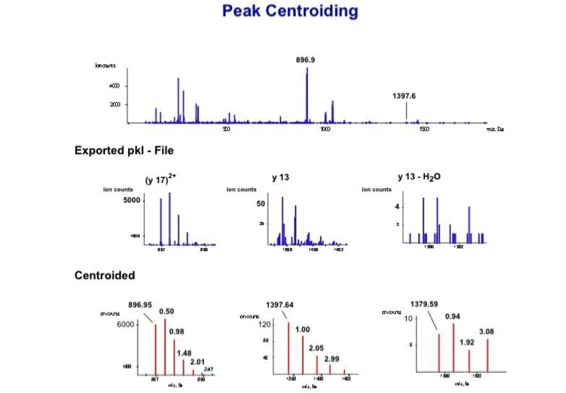
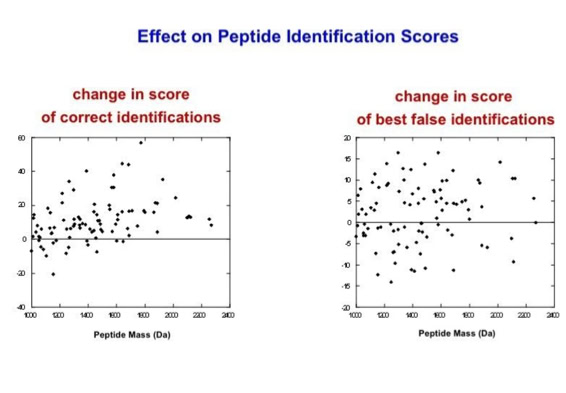
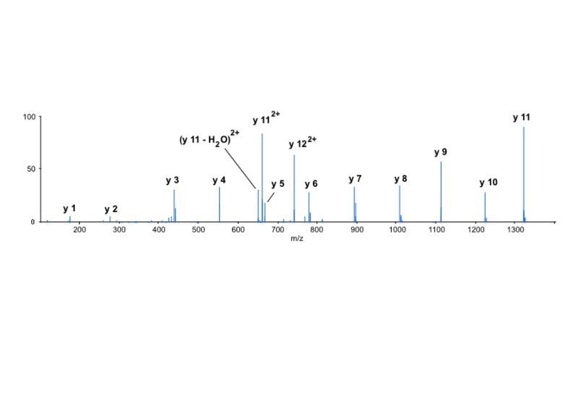
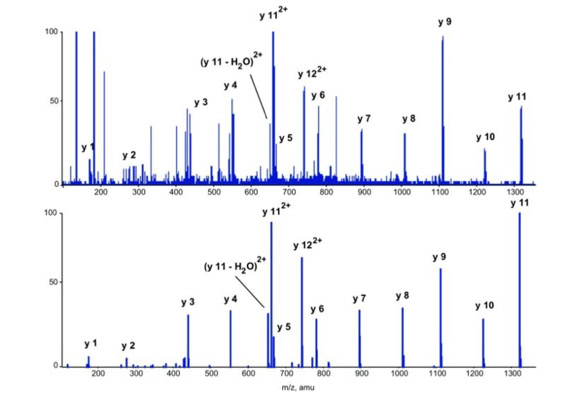
With the switch to automated data acquisition and peptide identification came the need to develop software algorithms that would help in generating the best possible specificity in data base identifications. We used a quadrupole time of flight instruments to acquire the data and Mascot as search engine. To support the identification process we pre-processed the fragment spectra (slide 7 - 10). This improved remarkably the score for correctly identified peptides whereas the score for wrongly assigned peptides stayed nearly the same (slide 11, 12) [2]. The processing was implemented in form of scripts using Igor Pro as processing engine. Later Matrix Science made a similar programme publicly accessible (Mascot Distiller).
Differential scanning to improve de novo sequencing:
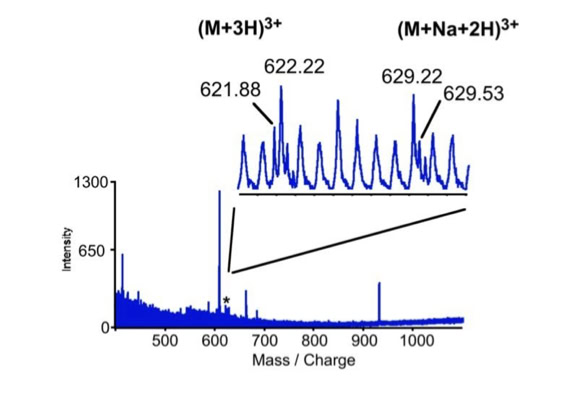
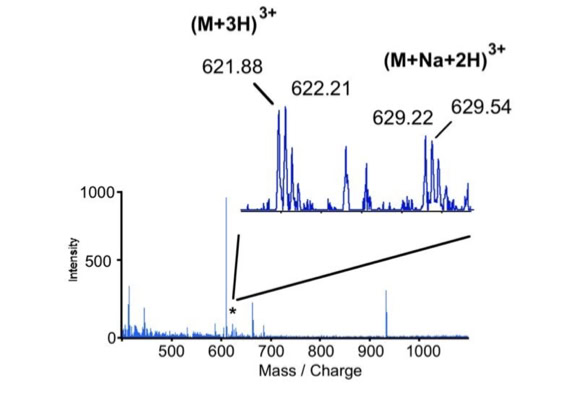
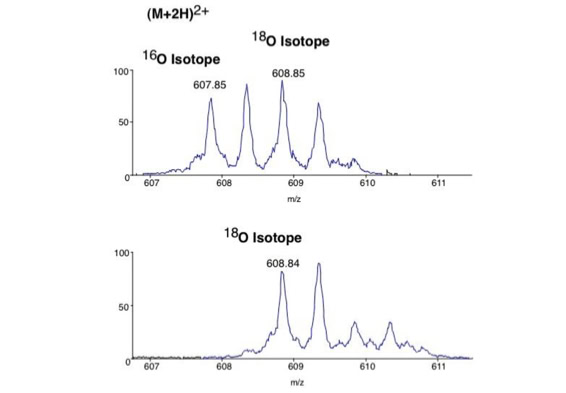
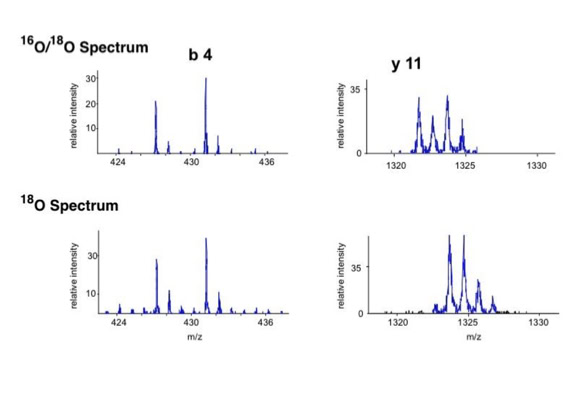
Differential scanning builds upon 18O labeling for de novo sequencing [3]. By digesting proteins in 1:1 18O/16O water all cleaved peptides carry to 50% an 18O isotope at their C-terminus. For the differential scanning method two fragment spectra per peptide are acquired, one including the entire isotopic envelope and a second with only the 18O isotopes selected (slide 13). When comparing these two fragment spectra C-terminal fragments differ by their isotopic representation (slide 15). This difference can be used by a software algorithm to generate a fragment spectrum filtered for C-terminal fragments (slide 16, 17) [4].
Parallel fragmentation of peptides to improve specific detection of secondary modifications:
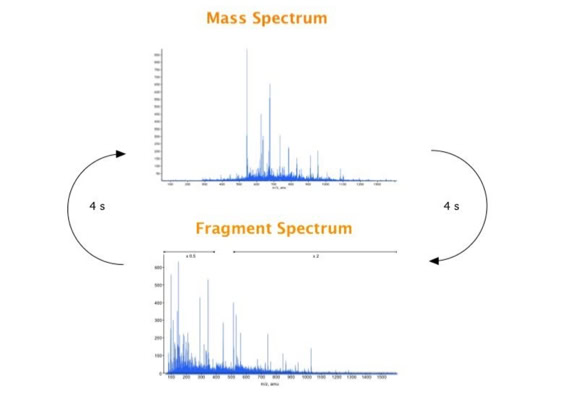
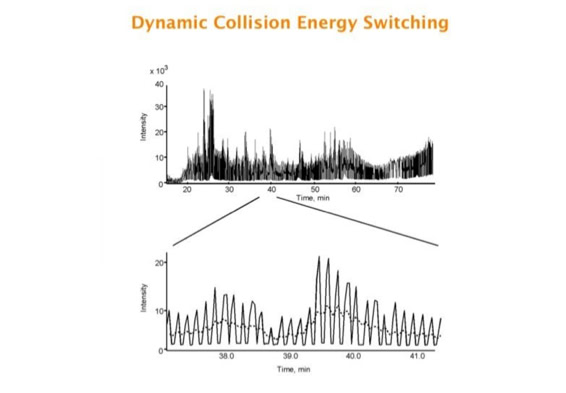
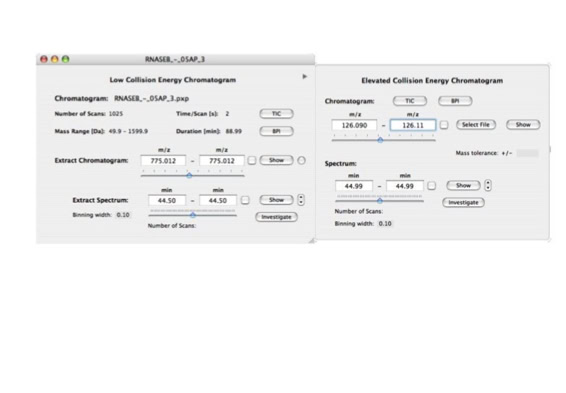
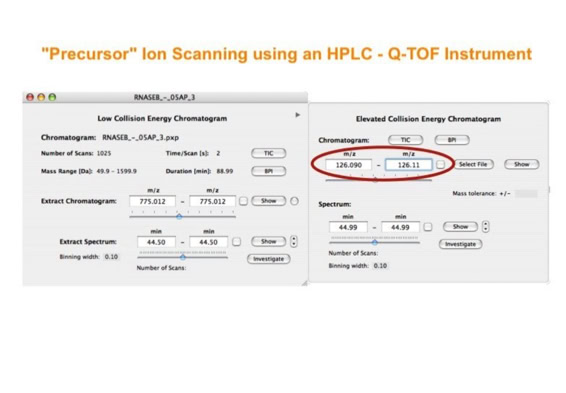
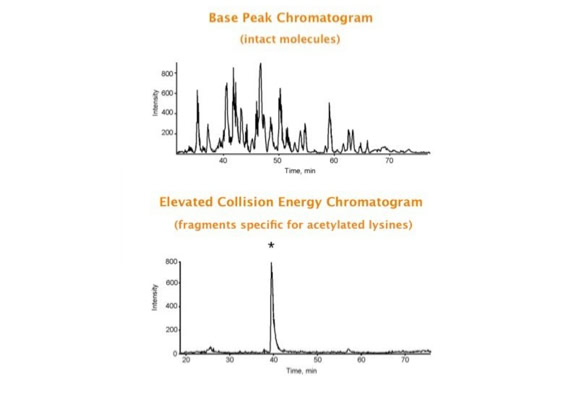
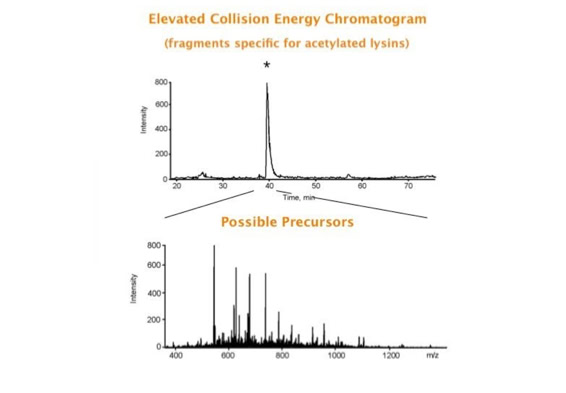
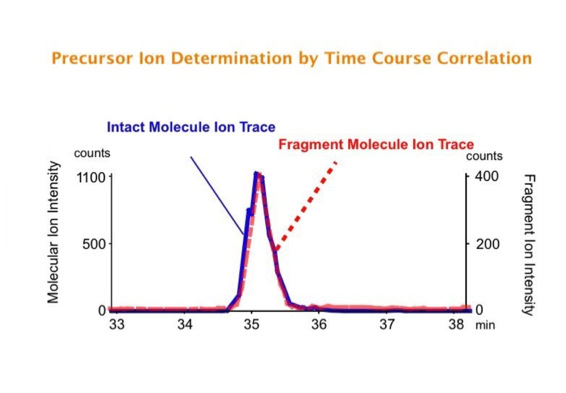
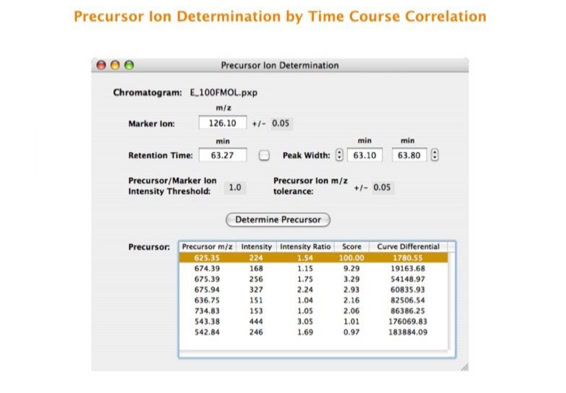
Parallel fragmentation of peptides is an attempt to increase the number of ions studied by their fragment spectrum. We explored this technique to acquire data that can be interrogated for many different marker ions or combinations of marker ions representing specific chemical structures like secondary peptide modifications. Instead of selecting an individual precursor the collision energy is permanently switched between an elevated and a low state. The mass spectrometer acquires intact masses and fragments of all the currently present molecules in an alternating way (slide 18, 19). Fragment data and intact molecular mass data can be separated by appropriate software tools (slide 20). Once the fragment data are isolated they can be interrogated for any marker ion trace or any combination of marker ions (slide 18). Once a marker ion specific for a peptide modification has been detected the true precursor that generated this marker ion upon fragmentation can be identified amongst all present precursors (slide 23) by correlating its chromatographic time profile to the time profile of the fragment (slide 24, 25). The real sequence of the precursor ion can be determined by running the same sample a second time and specifically select the highly scored precursors for fragmentation ignoring all the others [5].
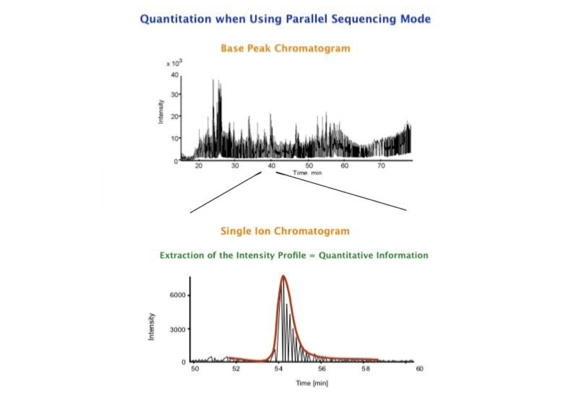
The fast switching of the mass spectrometer allows to reconstruct all chromatograms of all primary ions. The complete chromatogram gives access to their total ion volume and with it to their quantity (slide 26) [6].
The time course correlation is one of the critical steps in this analytical strategy. New equipment increases this time resolution - either by an improved LC separation using ultra high pressure chromatography equipment (Waters) or by using ion drift instruments (Prof. Clemmer). Once the time resolution is of a sufficient degree entire fragment spectra can be reconstructed for the observed precursors (parallel sequencing). The second LC run becomes superfluous.
The advantages of parallel sequencing for the analysis of very complex mixtures can be considerable. As demonstrated by scientists at Waters, qualitative and quantitative information can be generated on a systematic basis from every analysis. Prof. Clemmer's group demonstrated that it is possible to analyze proteomic samples much more rapidly covering a large number of peptides [7-10]. Since all masses are measured with time of flight instruments the high mass accuracy allows sufficiently accurate database searches.
References*
* It is not intended that the reference list reflects the entire research in the field.
Sequence & Identify Proteins
In 1992 a mass spectrometry group was established under the leadership of Matthias Mann. In the following years we explored how to sequence peptides and identify proteins using mass spectrometers in the most sensitive way. There were three important components to this
• the nano-electrospray ion source (figure 1 - 4)
• the sequence tag based protein identification algorithm (slide 5)
• protocols for gel staining, in-gel digestion and sample preparation for the analysis with mass spectrometry


Figure 1: Nano-Electrospray Emitter
Figure 2: Nano-Electrospray Ion Source
The nano-electrospray ion source:
A theoretical and experimental study of the electrospray process done in 1988 / 1989 at the University of Münster in the department of experimental physics in the group of Prof. Benninghoven (by Matthias Wilm) let to the construction principle of the nano electrospray ion source [1].
The result was that an electrospray emitter produces exclusively droplets with a diameter of 200 nm or less when operated at a flow rate of 20 nL/min or less. The spray should be generated purely electrostatically emitting the droplets from a stable Taylor Cone. The small size is so important because every droplet contains on the average only one analyte molecule per droplet if the analyte concentration is in the order of 1 pmol/µl. The nano electrospray ion source could therefore represent an electrospray ion source with a 100% ionization efficiency [1, 2]. At its time these findings proposed a mechanism for the electrospray ionization effect itself. The molecules would be passively released into the vacuum system after evaporation of the solvent. They allowed the prediction that the electrospray ionization mechanism is a very soft mechanism and that it should be possible to ionize molecules or molecular assemblies of virtually unlimited mass. The experimental results at that time were already produced by an electrospray emitter made from pulled glass capillaries.
The hoped for characteristics of the later called nano-electrospray ion source could be realized in the early 90's at the EMBL in the group of Matthias Mann. The pulled glass capillaries are the key element of the ion source. They were produced with a micro-capillary puller. Their surface is covered with gold or any other metal to make them conductive. The needles are mounted in gas tight needle holder to allow the application of a small air pressure for supporting the constant flow of liquid through the narrow tip if required. When coming from the puller the orifice of the needles can be in the nanometers range. For stable operation they have to be opened to give an orifice of 1 - 2 µm by moving them gently against a surface under microscopic control. Since nano-electrospray needles when operated on flow rates of 10 - 20 nL / min produce exclusively very small droplets they can be directly interfaced with the vacuum system of a mass spectrometer. The short distance to the orifice of the mass spectrometer, its high ionization efficiency and the low flow rate makes the nano-electrospray source a very useful emitter for the analysis of individual samples for direct mass measurements or tandem mass spectrometric investigations [2].
The nano electrospray source had been made commercially available for a variety of instruments by Proxeon.


Figure 3: Nano-Electrospray Emitter
Figure 4: Puller Equipment to Generate Nano-Electrospray Emitters
Sequence tag based protein identification:
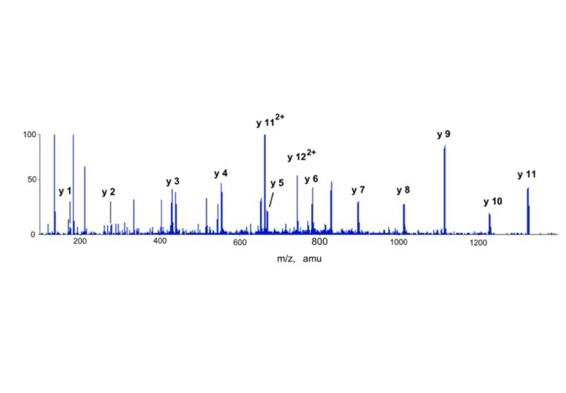
The sequence tag based protein identification algorithm is a manual way to identify protein based on the tandem mass spectrum of one or several of its peptides [3]. The algorithm was invented by Matthias Mann at a time when low resolved triple quadrupole fragment spectra were the only source of sensitive fragment spectra of peptides. A sequence tag is a short stretch of sequence read out from a fragment spectrum of a peptide.
In the example of slide 5 which shows the fragment spectrum of the peptide TTPAVLDSDGSYFLYSK the sequence tag is (1079.61)SD(IL)(1394.80). The amino acids I and L can not be distinguished in a low energy fragment spectrum. To lead to the correct identification of the underlying peptide the sequence tag has to connect a continuous series of b or (exclusive or) y ions. If a peptide had been found via a database search the complete peptide sequence is compared with the complete fragment spectrum to find other sequence specific ions for confirming the identification. The sequence tag based identification algorithm is a very specific identification method.
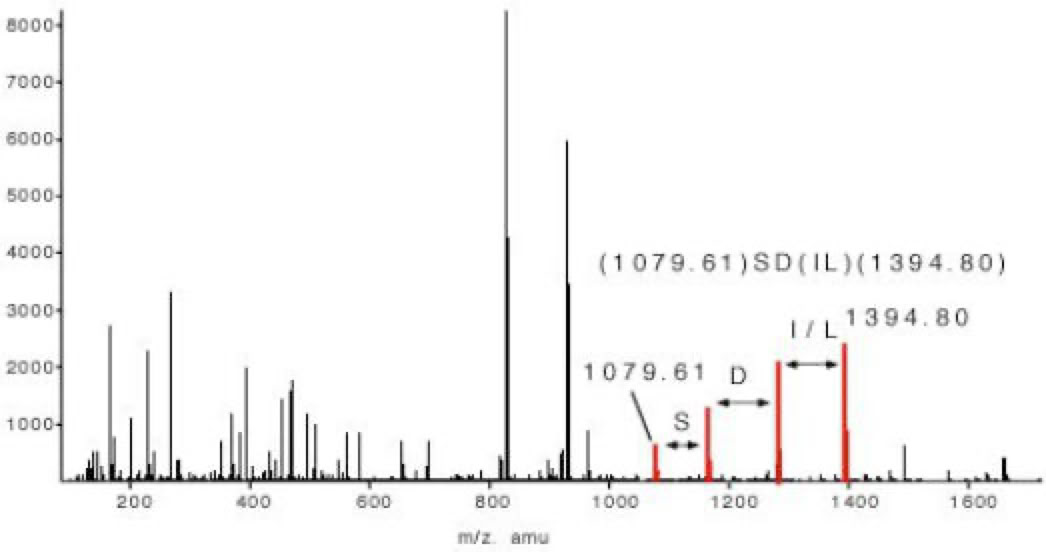
Figure 5: Sequence Tag
Sample preparation protocols:
Several protocols relevant for the analysis of proteins by mass spectrometry had been used in our group. The silver staining and the in-gel digestion protocol had been developed by Andrej and Anna Shevchenko [4].
• Mass spectrometry compatible silver staining of gels: silver staining protocol
• In-gel digestion of proteins to be analyzed by mass spectrometry: in-gel digestion protocol
• Two methods for the sample preparation for MALDI: dried droplet, fast evaporation protocol
• A selection of MALDI matrices: MALDI matrices
By introducing 18O labelling it became possible to sequence long amino acid stretches from peptide fragment spectra de novo [5].
References*
* It is not intended that the reference list reflects the entire research in the field.